
Ricerca scientifica
Aprile 7, 2014Neoplasie intraduttali papillari mucinose (IPMN)
Ottobre 8, 2019Le neoplasie neuroendocrine del pancreas sono un gruppo eterogeneo di lesioni che originano dalla componente endocrina del pancreas stesso. Sono relativamente rare e differiscono dal carcinoma del pancreas (con questo intendiamo adenocarcinoma del pancreas) per tre aspetti. In primo luogo,le neoplasie endocrine hanno una crescita generalmente molto lenta rispetto al cancro del pancreas e quindi i pazienti con tumore endocrino hanno una prognosi migliore rispetto ai pazienti con adenocarcinoma del pancreas. In secondo luogo, alcuni tumori neuroendocrini producono grandi quantità di ormoni specifici e il cui rilascio può provocare quadri clinici con sintomi drammatici. Ad esempio, alcuni tumori neuroendocrini chiamati “insulinomi” producono grandi quantità di insulina che viene rilasciata nel flusso sanguigno causando un drammatico abbassamento dei livelli di zucchero nel sangue (chiamato ipoglicemia). In terzo luogo, alcune neoplasie endocrine si presentano in pazienti con una sindrome genetica familiare. Ad esempio, i pazienti affetti da Neoplasia endocrina multipla di tipo 1 (MEN 1), causata da una mutazione ereditaria nel gene MENIN localizzato sul cromosoma 11, sviluppano tumori ipofisari, tumori della loro ghiandola paratiroidea e tumori neuroendocrini del pancreas. La valutazione del rischio familiare ed il counseling genetico è fondamentale per l’individuazione precoce della malattia.
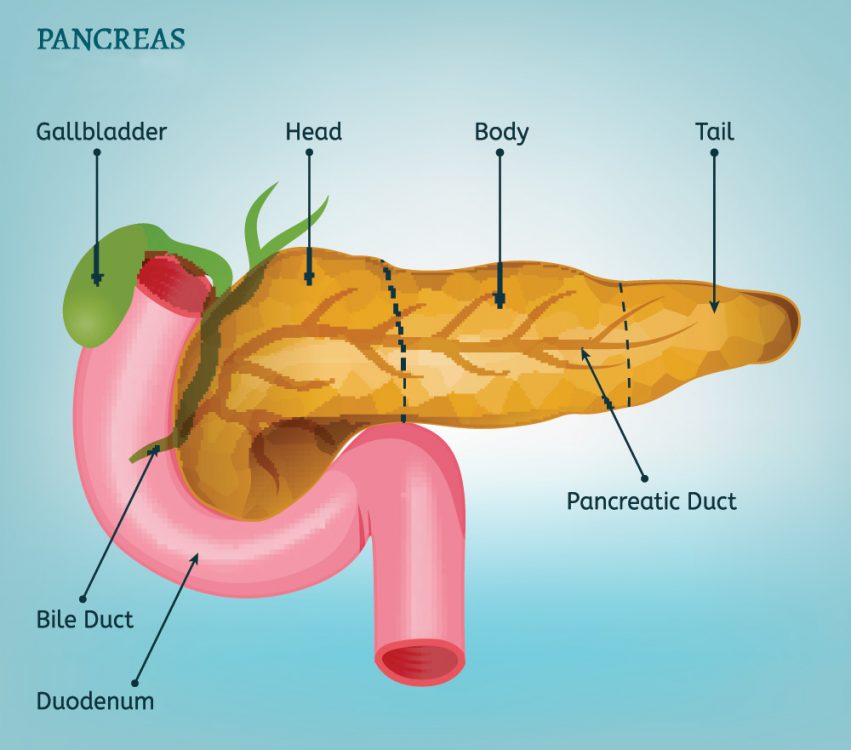
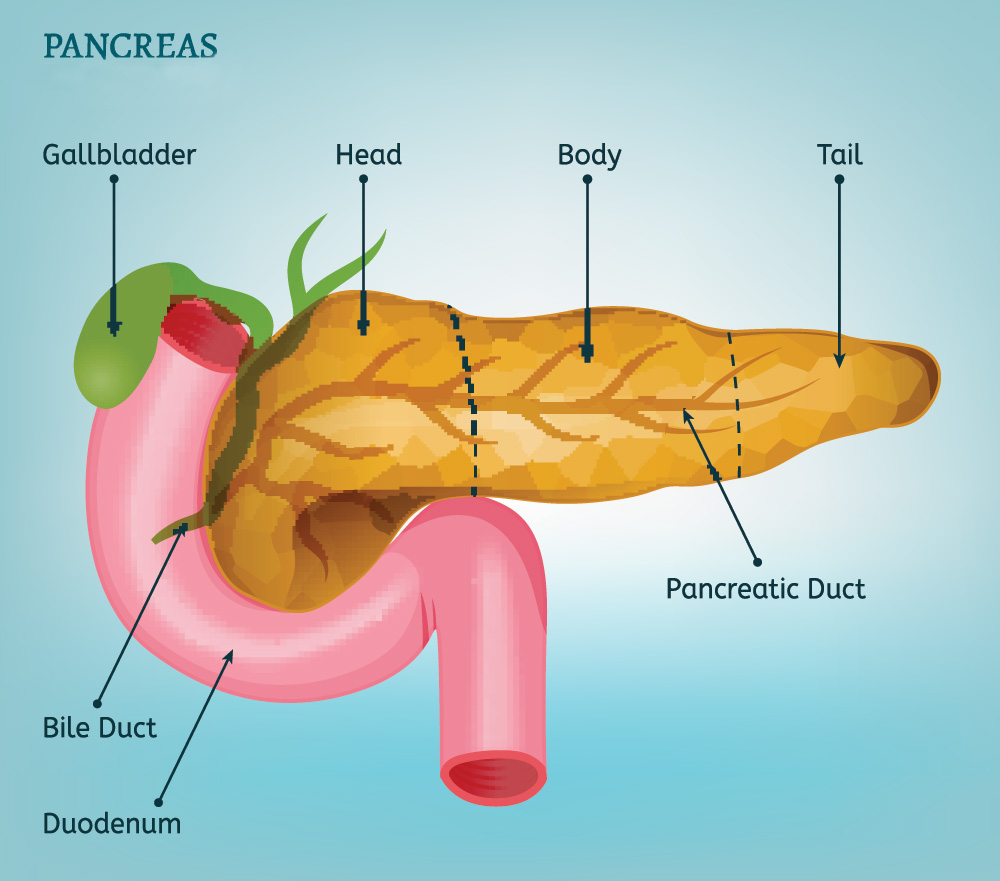
Il pancreas è funzionalmente diviso in due parti. La maggior parte del pancreas produce gli enzimi per la digestione del cibo. Questa porzione del pancreas è il pancreas “esocrino”: è da questa parte che origina il tumore al pancreas chiamato adenocarcinoma duttale. Il pancreas contiene poi numerose isole di cellule endocrine chiamate “isole di Langerhans”. Le cellule endocrine contenute in queste isole hanno il compito di produrre ormoni che controllano i livelli di zucchero nel sangue, ovvero insulina, glucagone e somatostatina. L’insulina abbassa i livelli di zucchero nel sangue e il glucagone generalmente aumenta i livelli di zucchero nel sangue. Le neoplasie endocrine del pancreas sono divise in “funzionanti” quando producono sintomi perché rilasciano ormoni nel flusso sanguigno e “non funzionanti” quando non rilasciando ormoni nel flusso sanguigno non danno sintomi ma determinano solo un effetto massa. In passato la maggior parte delle neoplasie endocrine pancreatiche scoperte clinicamente erano funzionanti mentre oggi, con il progresso delle tecniche di imaging, viene diagnosticato un numero maggiore di neoplasie endocrine pancreatiche prevalentemente non funzionanti e determinanti esclusivamente un effetto massa sul pancreas. Convenzionalmente, i tumori endocrini pancreatici sono denominati in base alla loro sindrome clinica predominante e al prodotto ormonale: gli “insulinomi “ sono neoplasie endocrine del pancreas che producono insulina e determinano bassi livelli di zucchero nel sangue.
CAUSE
Tra le cause predisponenti alle neoplasie endocrine vi sono diverse sindromi genetiche (familiari). E’ importante riconoscere questi quadri poichè, trattandosi di sindrome genetiche causate da mutazioni genetiche trasmissibile e quindi ereditabili, altri membri della famiglia possono essere a rischio. In secondo luogo, i pazienti che hanno queste sindromi genetiche sono a maggior rischio di sviluppare uno o più tumori dell’apparato neuroendocrini, sia in sede pancreatica che extra-pancreatica.
La Neoplasia endocrina multipla di tipo 1, abbreviata in MEN-1, è una sindrome familiare causata da mutazioni ereditarie nel gene MEN1 (che codifica la proteina meninica) sul cromosoma 11. I pazienti con mutazioni del gene MEN-1 sono predisposti a sviluppare tumori dell’ipofisi, delle paratiroidi (localizzate nel collo ed importanti nel controllo dei livelli di calcio nel sangue) e del pancreas.
La sindrome di von Hippel-Lindau, abbreviata in VHL, è una sindrome familiare causata da mutazioni ereditarie del gene VHL sul cromosoma 3. I pazienti con von Hippel-Lindau sono predisposti allo sviluppo di tumori in un certo numero di organi tra cui il cervello (emangioblastoma), l’occhio (emangioblastoma), il rene (carcinoma a cellule chiare) e le ghiandole surrenali (feocromocitoma). Il tumore neuroendocrino del pancreas può essere la prima manifestazione di VHL e la maggior parte dei pazienti con VHL alla fine sviluppa un tumore al pancreas.
La sclerosi tuberosa complessa (TSC) è una terza sindrome genetica che predispone alle neoplasie neuroendocrine del pancreas. La sclerosi tuberosa è causata da mutazioni ereditarie in uno dei due geni: TSC1 o TSC2.
La neurofibromatosi di tipo 1, nota anche come malattia di von Recklinghausen, è una sindrome familiare causata da mutazioni ereditarie del gene NF1 sul cromosoma 17.
TEST EMATICI
I test ematici specifici (test immunologici e radioimmunologici) permettono di evidenziare livelli elevati di ormoni prodotti dalle neoplasie endocrine. Idosaggi di insulina, gastrina, VIP, glucagone, somatostatina, polipeptide pancreatico, prostaglandine e altri marcatori ormonali vengono in genere eseguiti presso grandi centri medici o laboratori di riferimento selezionati.
LOCALIZZAZIONE E STADIAZIONE
Al momento, l’esame diagnostico iniziale raccomandato per la localizzazione delle neoplasie endocrine del pancreas è la TAC con contrasto endovenoso e orale. L’accuratezza della TAC nella localizzazione del tumore è migliorata dall’uso del contrasto sia orale che endovenoso. La TAC consente l’accurata valutazione di eventuali linfoadenomegalie peripancreatiche e la presenza di metastasi epatiche. Le neoplasie endocrine del pancreas in genere producono masse solide alla scansione TC e si impregnano di mezzo di contrasto (“enhancement”) in fase arteriosa. Si ritiene che quest’ultima caratteristica sia una manifestazione della ricca vascolarizzazione di questi tumori ed è una utile funzionalità diagnostica. L’esame angiografico invasivo è una metodica diagnostica di secondo livello nel caso in cui il tumore primario non fosse rilevabile all’esame TAC, ed è basata sulla visualizzazione selettiva della vascolarizzazione arteriosa del pancreas e delle regioni peripancreatiche.
L’ ecoendoscopca rappresenta una risorsa diagnostica importante e permette di eseguire una biopsia.
L’altro importante esame diagnostico nello studio dei tumori endocrini del pancreas è l’Octreoscan. Questa tecnica si basa sulla presenza di recettori della somatostatina su molti tumori endocrini del pancreas e ha il potenziale di identificare sia i tumori primari che le localizzazioni secondarie epatiche ed extra-epatiche. Fondamentalmente, una forma radioattiva di octreotide, un farmaco simile alla somatostatina, viene utilizzata affinchè si leghi alle cellule dei tumori neuroendocrini del pancreas esprimenti sulla loro superficie il recettore per la somatostatina. Un dispositivo di misurazione delle radiazioni può quindi rilevare l’octreotide radioattivo localizzato nel tumore.
TRATTAMENTO CHIRURGICO
La resezione chirurgica è l’opzione curativa per i pazienti con neoplasie endocrine del pancreas. Al momento dell’esplorazione chirurgica della neoplasia endocrina del pancreas, viene eseguita una valutazione completa del pancreas e delle regioni peripancreatiche. La tecnica che fornisce ulteriori informazioni è l’ecografia intraoperatoria, che può aiutare nell’identificazione del tumore. In generale, i chirurghi possono eseguire una pancreaticoduodenectomia secondo Whipple o con preservazione del piloro per i tumori della testa del pancreas, e una pancreatectomia distale con o senza preservazione della milza per i tumori della coda del pancreas. Gli obiettivi della terapia chirurgica per le neoplasie endocrine del pancreas includono: il controllo dei sintomi riconducibile alla secrezione ormonale, la resezione oncologicamente radicale della massa tumorale e la conservazione del parenchima pancreatico massimo.
TRATTAMENTO MEDICO
A causa del basso tasso di replicazione cellulare le neoplasia neuroendocrine non sono candidabili alla chemioterapia convenzionale (che di solito insiste su cellule a rapida replicazione). Recentemente la Food and Drug Administration (FDA) degli Stati Uniti ha recentemente approvato i farmaci Sutent (sunitinib) e Afinitor (Everolimus) per il trattamento dei tumori endocrini pancreatici avanzati (noti anche come tumori a cellule di isole o tumori neuroendocrini del pancreas. Everolimus (inibitore di mTOR) è stato approvato per il trattamento di pazienti con tumori neuroendocrini pancreatici progressivi che non sono resecabili chirurgicamente, localmente avanzati o metastatici. Il trattamento con Sunitinib ha dimostrato di migliorare significativamente la sopravvivenza libera da progressione in pazienti con tumori neuroendocrini pancreatici metastatici. In casi selezionati nei pazienti metastatici, le lesioni epatiche possono essere trattate bloccando il loro apporto di sangue usando tecniche come l’embolizzazione dell’arteria epatica e la chemioembolizzazione.
PROGNOSI
Il fattore prognostico più importante è la possibilità di resecare chirurgicamente il tumore. Altri fattori prognostici comprendono le dimensioni del tumore, la presenza o l’assenza di invasione dei vasi sanguigni, la presenza o l’assenza di metastasi ai linfonodi o altri organi. I tassi di sopravvivenza a 5 anni oscillano tra il 50 e il 70% nella maggior parte delle casistiche presenti in letteratura.
INSULINOMA
L’insulinoma rappresenta la neoplasia endocrina pancreatica più comune. La sindrome da insulinoma è caratterizzata dalla “triade di Whipple:” 1) sintomi di ipoglicemia (bassi livelli di zucchero nel sangue) durante il digiuno, 2) evidenza laboratoristica di ipoglicemia con glicemia (zucchero) inferiore a 50 mg / dl e 3) sollievo dei sintomi dopo somministrazione di glucosio. I sintomi includono confusione, convulsioni, ottundimento, alterazioni della personalità e coma, nonché palpitazioni, tremori, diaforesi (sudorazione) e tachicardia (battito cardiaco accelerato). Nella maggior parte dei casi, i pazienti consumano pasti e spuntini ricchi di carboidrati per alleviare o prevenire questi sintomi. L’insulinoma viene diagnosticato in modo più affidabile monitorando clinicamente e laboratoristicamente il paziente durante un digiuno prolungato. Durante il digiuno, il sangue viene campionato ogni 4-6 ore e al momento della comparsa della sintomatologia per determinare il glucosio e l’insulina. Un ulteriore supporto alla diagnosi di insulinoma deriva dal calcolo del rapporto insulina / glucosio in diversi punti temporali durante il digiuno monitorato. Gli individui normali avranno rapporti I:G inferiori a 0,3, mentre i pazienti con insulinoma mostrano in genere rapporti I:G superiori a 0,4 dopo un digiuno prolungato. Per l’insulinoma gli studi di imaging standard includono TC addominale, ecografia endoscopica ed angiografia viscerale. Il trattamento dell’insulinoma è chirurgico in quasi tutti i casi. Gli insulinomi si trovano uniformemente distribuiti all’interno del pancreas, con circa un terzo localizzato nella testa e processo uncinato del pancreas, un terzo nel corpo della ghiandola e un terzo nella coda pancreatica. Nel 90% dei pazienti si riscontra piccoli tumori solitari suscettibili di guarigione chirurgica. Il 10% dei pazienti con insulinoma presenta una sindrome della neoplasia-1 endocrina multipla (MEN-1). Il 10% di tutti i casi si presenta alla diagnosi con localizzazioni metastatiche ai linfonodi peripancreatici o al fegato, configurando il quadro clinico di insulinoma maligno. In queste circostanze, è possibile prendere in considerazione la resezione chirurgica del tumore primitivo e delle metastasi aggredibili. Il “debulking” del tumore può essere utile nel ridurre i sintomi ipoglicemici che possono minacciare la sopravvivenza a lungo termine. In media il paziente sopravvive diversi anni dopo la diagnosi ed il trattamento degli insulinomi maligni, indicando che la storia naturale di questi tumori è generalmente indolente. Il trattamento medico si giova dei seguenti agenti terapeutici: streptozocina, dacarbazina, doxorubicina e 5-fluorouracile. I più alti tassi di risposta alla chemioterapia sono stati osservati usando la terapia di combinazione.
GASTRINOMA (SINDROME DI ZOLLINGER-ELLISON)
I gastrinomi sono neoplasie endocrine del pancreas caratterizzate dal rilascio di grandi quantità di ormone gastrina ed una importante sindrome da iperacidità gastrica e duodenale. Nel 1955 Zollinger ed Ellison descrissero due pazienti con grave ulcera peptica e tumori endocrini del pancreas, postulando la causa risiedesse in un ormone ulcerogenico prodotto dal pancreas. Attualmente si stima che un paziente su 1000 con ulcera duodenale primaria e due pazienti su 100 con ulcera recidive post gastrectomia sono affetti da gastrinoma. Il 75% dei gastrinomi sono sporadici, mentre il 25% è associato alla sindrome MEN-1. In passato, la maggior parte dei gastrinomi veniva diagnosticata in fase metastatica. Più recentemente, grazie alla maggiore consapevolezza ed allo screening precoce, la diagnosi di gastrinoma viene condotta precocemente, portando alla scoperta di una percentuale più alta di neoplasie curabili. Clinicamente fino al 90% dei pazienti mostra una ulcerazione peptica del tratto gastrointestinale superiore e dolore addominale. Il 50% dei pazienti ha un certo grado di diarrea, mentre circa il 10% dei pazienti presenta diarrea come sintomo solitario. La diagnosi di gastrinoma deve essere sempre sospettata in questi contesti clinici incoraggiando il dosaggio della gastrinemia come strumento di screening. L’analisi dell’acido gastrico è un test importante nella valutazione di pazienti con sospetto gastrinoma, in quanto può distinguere tra cause ulcerogeniche (acido gastrico elevato) di ipergastrinemia e cause non ulcerogeniche (acido gastrico basso) di ipergastrinemia.
VIPOMA (SINDROME DI VERNER MORRISON)
I VIPomi sono neoplasie endocrine pancreatiche che causano sintomi rilasciando grandi quantità dell’ormone VIP nel flusso sanguigno. I pazienti caratteristicamente presentano diarrea grave intermittente, tipicamente di natura acquosa, con una media di 5 litri al giorno (che è una terribile diarrea!). L’ipopotassiemia (basso livello di potassio nel sangue) deriva dalla perdita fecale di grandi quantità di potassio (fino a 400 meq / giorno) e bassi livelli di potassio nel sangue possono essere associati a debolezza muscolare, letargia e nausea.
GLUCAGONOMA
I glucagonomi sono neoplasie endocrine pancreatiche che causano sintomi rilasciando grandi quantità di ormone glucagone nel flusso sanguigno. I sintomi più comuni nella sindrome del glucagonoma includono dermatite grave (eruzione cutanea), diabete lieve, stomatite (afte), anemia (basso numero di globuli rossi) e perdita di peso. La dermatite si manifesta con una caratteristica eruzione cutanea chiamata “eritema migrante necrolitico“. Questa eruzione mostra migrazioni cicliche con chiazze eritematose (rosse) che si diffondono con punti di guarigione centrali.
SOMATOTATINOMA
I somatostatinomi sono neoplasie endocrine pancreatiche che causano sintomi rilasciando grandi quantità di ormone somatostatina nel flusso sanguigno. Il somatostatinoma è una neoplasia rara e presenta un’incidenza annuale stimata inferiore a una su quaranta milioni di persone. Le caratteristiche cliniche della sindrome del somatostatinoma non sono specifiche e comprendono la steatorrea (feci oleose), il diabete, l’ipocloridria (bassi livelli di cloruro nel sangue) e la colelitiasi (calcoli nella cistifellea). La diagnosi è clinica e biochimica (livelli elevati di somatostatina). La maggior parte dei somatostatinomi è stata localizzata nella testa del pancreas.
NEOPLASIE NEUROENDOCRINE NON FUNZIONANTI
La maggioranza dei pazienti con neoplasie endocrine pancreatiche non presenta una sindrome clinica causata dalla produzione in eccesso di ormoni. Le manifestazioni cliniche più frequenti sono: dolore addominale, perdita di peso ed ittero, risultanti da lesioni occupanti spazio nel pancreas. Frequente è il riscontro occasionale in corso di esami TAC o RMN addome eseguiti per altre patologie. Le neoplasie endocrine del pancreas non funzionanti si trovano più comunemente nella testa, nel collo o nel processo unicinato del pancreas. Una parte di queste neoplasia è biologicamente maligna. Tuttavia, contrariamente alla scarsa prognosi associata all’adenocarcinoma duttale questi tumori neuroendocrini non funzionanti tendono a crescere in modo più indolente e sono associati a una sopravvivenza più lunga. La TAC addominale viene utilizzata per la valutazione del tumore primario e per la valutazione delle metastasi epatiche. All’intervento chirurgico la maggior parte di queste neoplasie non funzionali sono più grandi di 2 cm e richiedono per i tumori nella testa, del collo o nel processo uncinato del pancreas un intervento di pancreaticoduodenectomia (resezione di Whipple), mentre i tumori che si presentano nel corpo o nella coda del pancreas sono trattati con pancreatectomia distale. Il tasso complessivo di sopravvivenza a 5 anni in tutti i pazienti con neoplasie pancreatiche non funzionali resecate si avvicina al 50%. Negli ultimi anni le forme incidentali(non funzionanti) sono aumentate grazie all’ampio utilizzo di tecniche d’imaging di alta qualità. Secondo le recenti linee guida per il trattamento dei tumori neuroendocrini, per le lesioni non funzionanti ≤ 2 cm e in assenza di sintomi e/o sospette lesioni metastatiche, si può adottare un trattamento conservativo arruolando il paziente in un programma di follow-up clinico e radiologico. Le neoplasie neuroendocrine non funzionanti con diametro ≤ 2 cm presentano un rischio di “malignità” attorno al 6% mentre la mortalità per malattia a 5 anni è nulla. Non esiste un programma di follow-up basato sull’evidenza; di solito viene suggerito un controllo clinico e radiologico annuale e dopo sei mesi dalla prima diagnosi. Per quanto riguarda neoplasie non funzionanti > 2 cm l’approccio chirurgico rimane il trattamento di scelta.
{kind=link}
{kind=link}